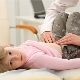
Atrofia muscular espinal en niños

La atrofia muscular espinal es una patología grave que a menudo realiza acciones simples, como caminar, sentarse, inaccesible para un niño. El bebé puede incluso ser privado de una oportunidad tan natural como la respiración independiente. Es muy difícil hacer predicciones para esta patología, Después de todo, no existe un tratamiento especial ni profilaxis, pero todo depende de la forma de la aflicción y los factores., Qué medicamento no puede encontrar una explicación.
Que es esto
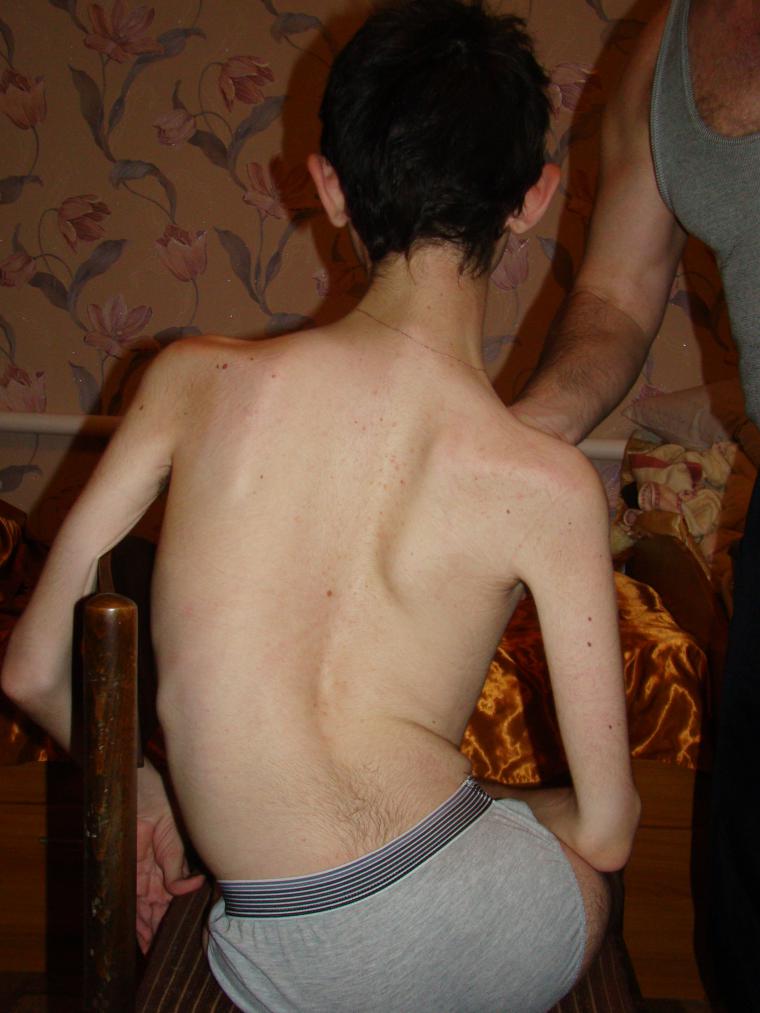
Hablando de atrofia muscular espinal, no implica una enfermedad específica, sino todo un grupo de enfermedades con la abreviatura general CMA. Todos ellos son hereditarios y están asociados con la degeneración de las células nerviosas de la médula espinal, que son responsables de las funciones motoras.
Entre los Las patologías genéticas en niños. Las atrofias musculares espinales ocupan los primeros lugares en términos de frecuencia de diseminación. Y aproximadamente uno de cada 6 mil niños nace con un diagnóstico tan terrible. En el 50% de los casos, los niños no viven hasta los dos años y mueren. El resto de la vida es una discapacidad.

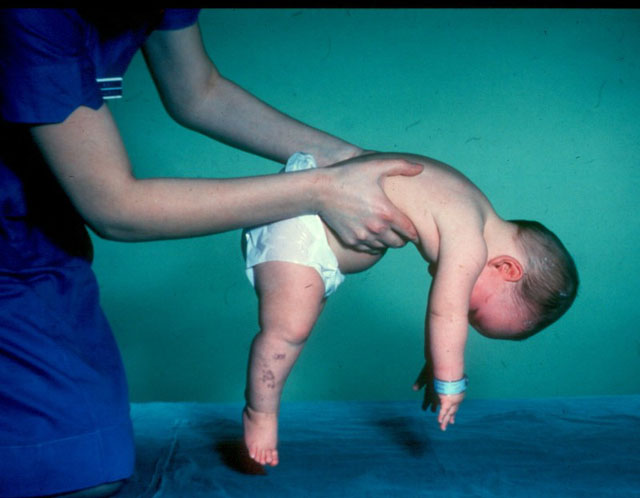
El problema, según los genetistas, es mucho más amplio de lo que parece a partir de las estadísticas proporcionadas.
La enfermedad se desarrolla debido a la mutación de ciertos genes, y uno de ellos es SMN1, que se considera el principal "culpable" de la patología, en cada forma modificada, cada habitante número cincuenta del planeta está presente en la recesión. Esto significa que los padres sanos, que ni siquiera se dan cuenta de que son portadores recesivos del gen mutado, pueden tener un bebé con atrofia muscular espinal.
El grupo de enfermedades fue descrito por primera vez en el siglo XIX por Guido Verding, cuyo nombre fue nombrado más tarde como una de las variedades infantiles de AGR.
Clasificación
La forma más común de SMA en niños es proximal. Está representado por varios tipos de enfermedades, y no todas se manifiestan inmediatamente después del nacimiento del niño.
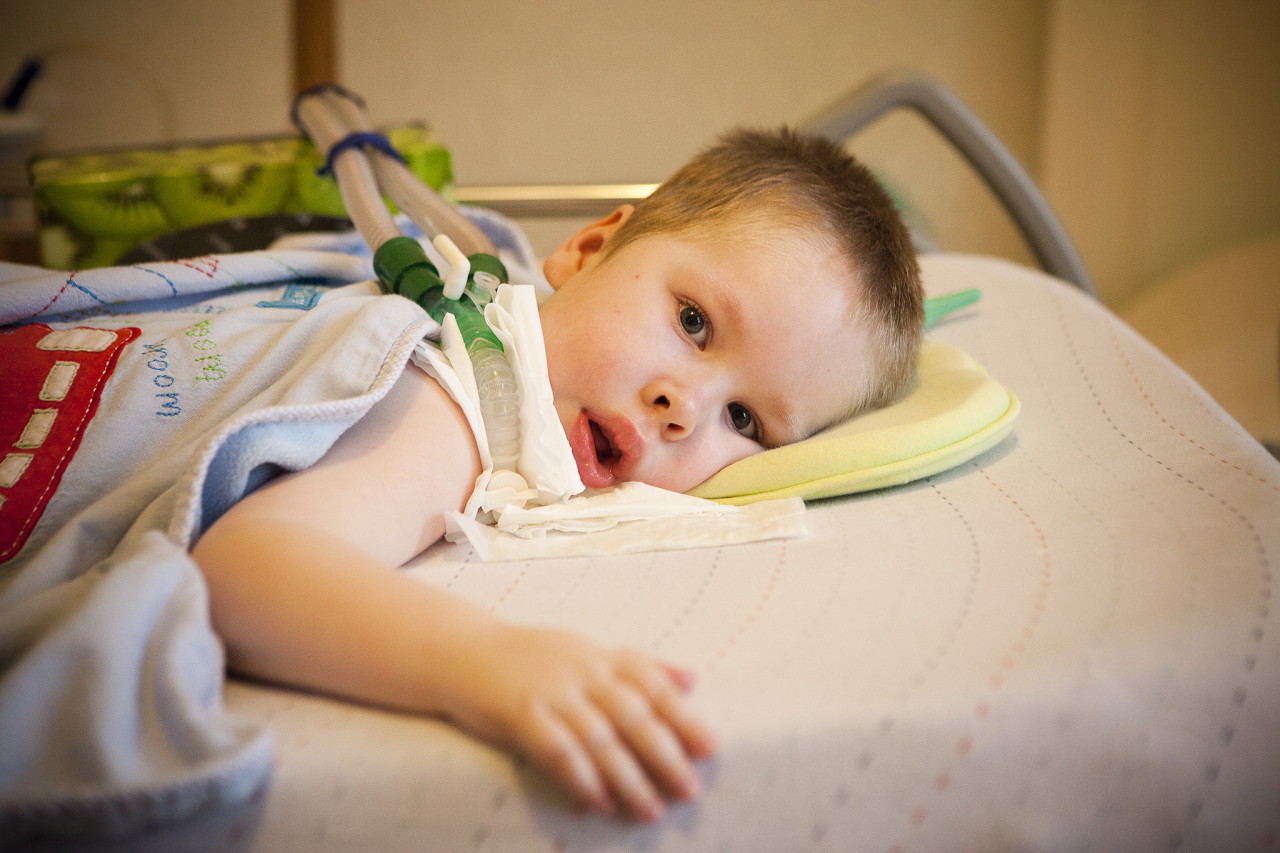
- Enfermedad de Verding-Hoffman - CMA tipo 1, enfermedad infantil graveque se manifiesta en los primeros seis meses de vida del niño. Las previsiones con la más desfavorable, la mayoría de los pacientes mueren. Un niño con SMA tipo 1 no puede ponerse de pie ni sentarse o rodar por sí solo. Muchos recién nacidos tienen alteración de los reflejos de succión y deglución. A menudo no hay posibilidad de respiración espontánea o la respiración es difícil.
- Atrofia Dubovitsa - CMA tipo 2, infante tardío Por lo general, se produce entre las edades de seis meses y un año y medio y más tarde. El niño no puede caminar, estar de pie, pero puede sentarse, la comida no se altera, se enfrenta a la tarea de tragar, chupar. El tiempo de vida del bebé depende del estado de los músculos respiratorios.
- Atrofia de Kugelberg-Welander - tipos de CMA 3, infantil Generalmente se encuentra en la edad de un año y medio, generalmente en dos años. Forma pronósticamente más favorable. Los pacientes pequeños pueden pararse, sentarse, moverse, pero experimentan una gran debilidad y, por lo tanto, en la mayoría de los casos necesitan una silla de ruedas, sin la cual su actividad vital normal es difícil para ellos.
- Atrofia de Kennedy - CMA tipo 4, bulbospinalnaya. Generalmente se considera una forma adulta, pero ocasionalmente se detecta en niños después de 15 años. La influencia de la vida rara vez se ve afectada, el debilitamiento de los músculos ocurre lentamente, gradualmente, una persona que llevó una vida normal y se consideró saludable, eventualmente se deshabilita y pierde la capacidad de moverse de forma independiente.


La atrofia de primera mano más o menos conocida de Duchenne y la atrofia de Vulpian es la SMA "adultos", el primero generalmente se detecta después de 18 años, y el segundo después de 20 años.
En los niños, no solo se registran formas aisladas de SMA, cuando nada le molesta excepto la distrofia muscular, sino también formas combinadas, cuando la atrofia espinal no es el único diagnóstico y el niño tiene otros problemas genéticos o congénitos, como defectos cardíacos y vasculares, oligofrenia.
Las razones
Como ya se mencionó, estamos hablando de una enfermedad genética y, por lo tanto, las razones de su aparición son el campo de búsqueda de genetistas. El niño hereda uno de los genes recesivos en el quinto cromosoma (estos pueden ser los genes SMN, NAIP, H4F5, BTF2p44).
La probabilidad de transmitir un gen de este tipo a una progenie desde un portador es alta: 25%. Si tanto la madre como el padre son portadores ocultos del gen mutado, entonces la probabilidad de que el niño sea AGR en un niño es del 50%. El gen anormal afectado previene la producción normal de proteína SMN y las células nerviosas responsables de las funciones motoras de los músculos de la médula espinal comienzan a morir gradualmente. El proceso de su muerte continúa incluso después de que nace el bebé.


Manifestaciones
Los síntomas dependen del tipo de enfermedad. Dado que consideramos solo cuatro tipos de niños, debe notarse que la debilidad muscular y la atrofia muscular son características de todos. De lo contrario, cada tipo tiene su propio cuadro clínico y características distintivas.
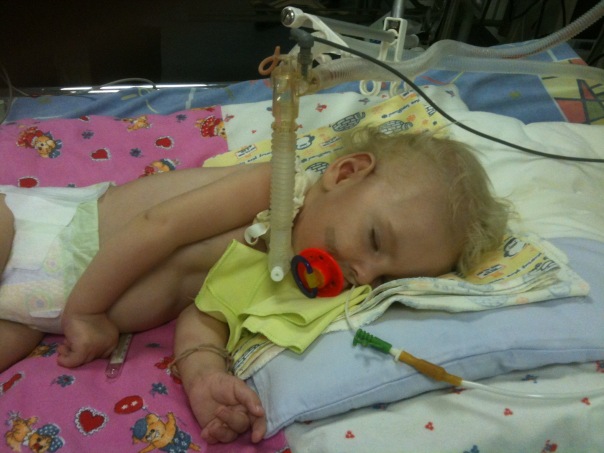
- CMA Tipo 1 (Atrofia de Verding-Hoffman) Disponible para la detección incluso durante el embarazo. El médico puede sospechar la enfermedad en el feto con agitación muy lenta. Pero para confirmar el diagnóstico en la etapa de llevar al niño es difícil, por lo general ocurre después del parto. Un niño con tal atrofia no puede sostener la cabeza por sí mismo, tirar de lado a lado, no se sienta. Casi siempre descansa sobre la espalda, su postura es relajada, no levanta las piernas, no las une, no pone las palmas juntas. En una etapa muy temprana, puede haber grandes problemas para alimentar al niño, ya que él se lo traga o no funciona. La mayoría de los niños mueren antes de los dos años. Algunos logran vivir hasta siete u ocho años, pero la atrofia solo aumenta. Por lo general, la muerte se produce debido a la insuficiencia del corazón, los pulmones, los órganos digestivos.
- Tipo CMA 2 (atrofia Dubovitsa) en el momento del nacimiento, generalmente no se detecta, porque el niño puede respirar, tragar alimentos y solo después de seis meses se hace evidente el progreso de la atrofia muscular. Si los primeros síntomas ocurren a la edad en que el niño ya ha aprendido a pararse en la cuna, entonces un signo cortante de las piernas, una gota irrazonable de migajas puede ser un signo brillante. Poco a poco, se vuelve difícil de tragar. Con el tiempo, el niño comienza a necesitar una silla de ruedas.
- Tipos de CMA 3 (amiotrofia de Kügelberg-Welander) Puede aparecer a cualquier edad después de los 2 años de edad. Un niño que normalmente creció y se desarrolló gradualmente comienza a quejarse de debilidad, generalmente en el área de los hombros, antebrazos. A medida que avanza, se le hace difícil correr, caminar escaleras, ponerse en cuclillas. Todo depende de la atención, algunos conservan la capacidad de moverse de forma independiente durante muchos años.
- CMA tipo 4 (atrofia de Kennedy) Se encuentra solo en pacientes masculinos, ya que se considera vinculado al cromosoma sexual X. Los primeros signos son debilidad en la región de los músculos del muslo, los nervios craneales se ven afectados gradualmente. La enfermedad progresa lentamente.

Tratamiento
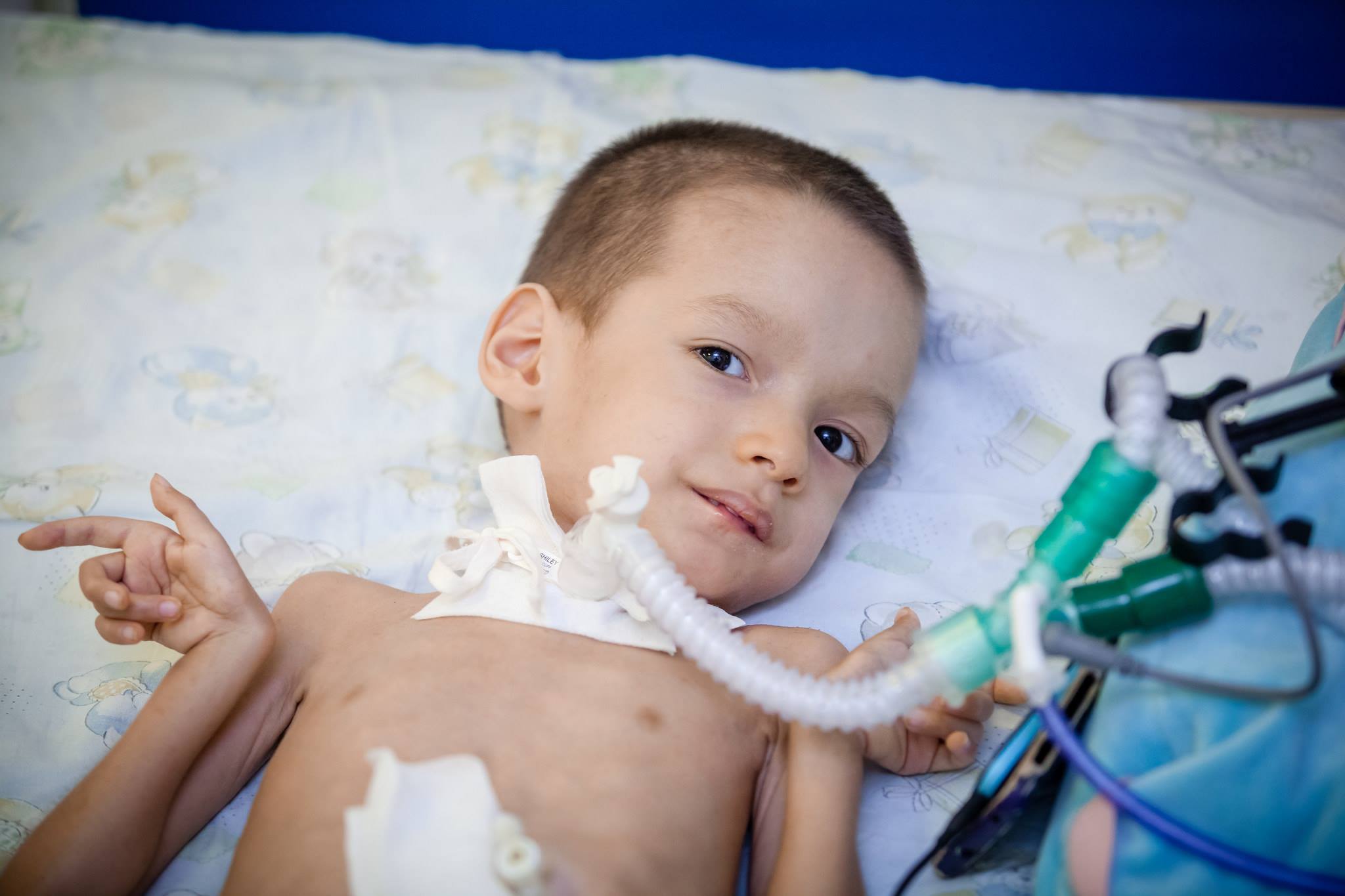
Desafortunadamente, hoy en día la ciencia médica no puede ofrecer métodos y medios para el tratamiento de SMA. No hay tales métodos. Para mantener las funciones del cuerpo y maximizar el período hasta que el niño pueda moverse, medicamentos como Prozerin, Oksazil. Reducen la actividad de una enzima capaz de escindir acetilcolina, que transmite un pulso de excitación a través de las fibras del sistema nervioso.
Tambien recomendado Uso sistemático de fármacos que aumentan el metabolismo energético a nivel celular, vitaminas del grupo B, fármacos nootrópicos, así como preparaciones de potasio y ácido nicotínico.
Un niño con SMA se muestra una dieta alta en proteínasPero estudios recientes han demostrado que el papel de la dieta es algo exagerado: no hay evidencia de que un alto contenido de proteínas en los alimentos afecte al menos de alguna manera la tasa de progresión de la enfermedad.
Pero con las calorías debe ser más cuidadoso, debido a la reducción de la actividad muscular, el niño puede ganar rápidamente kilos de más.
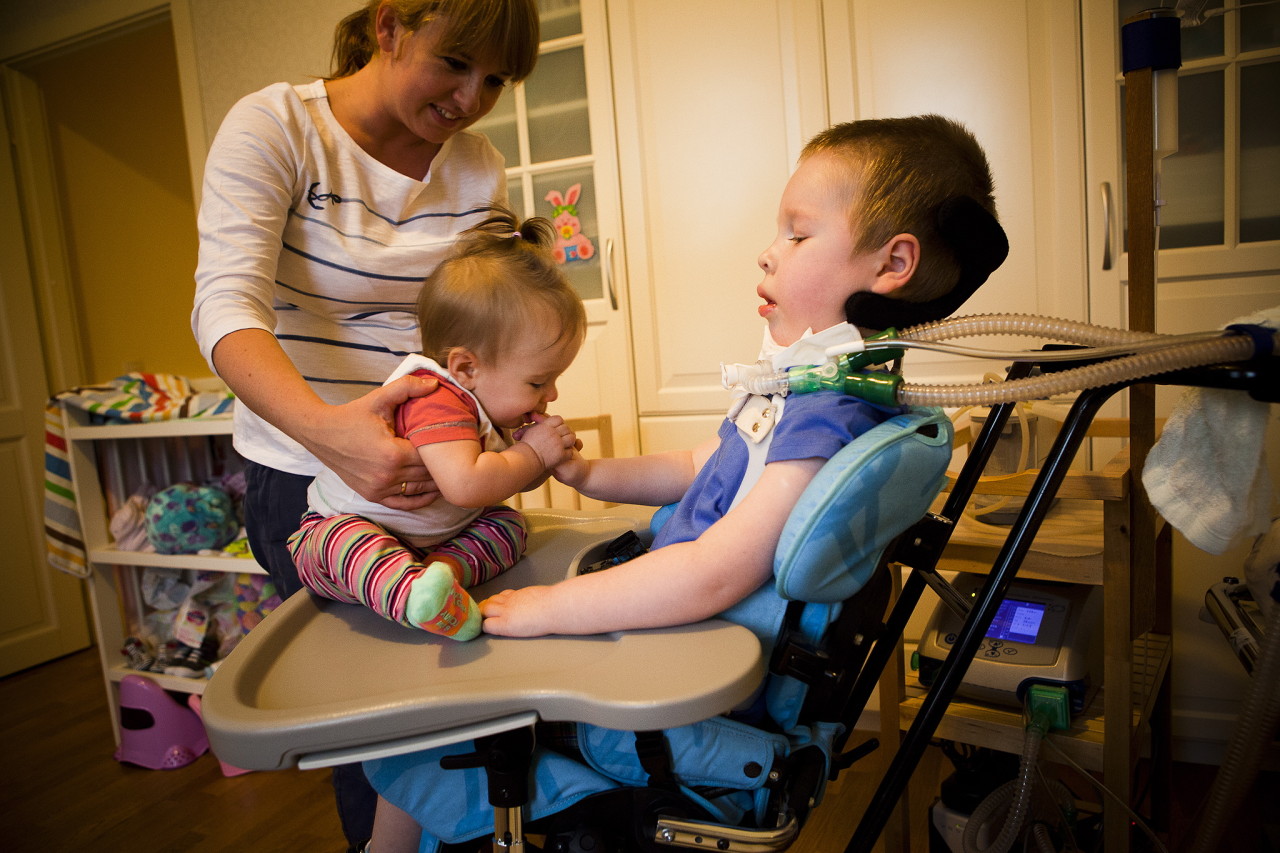
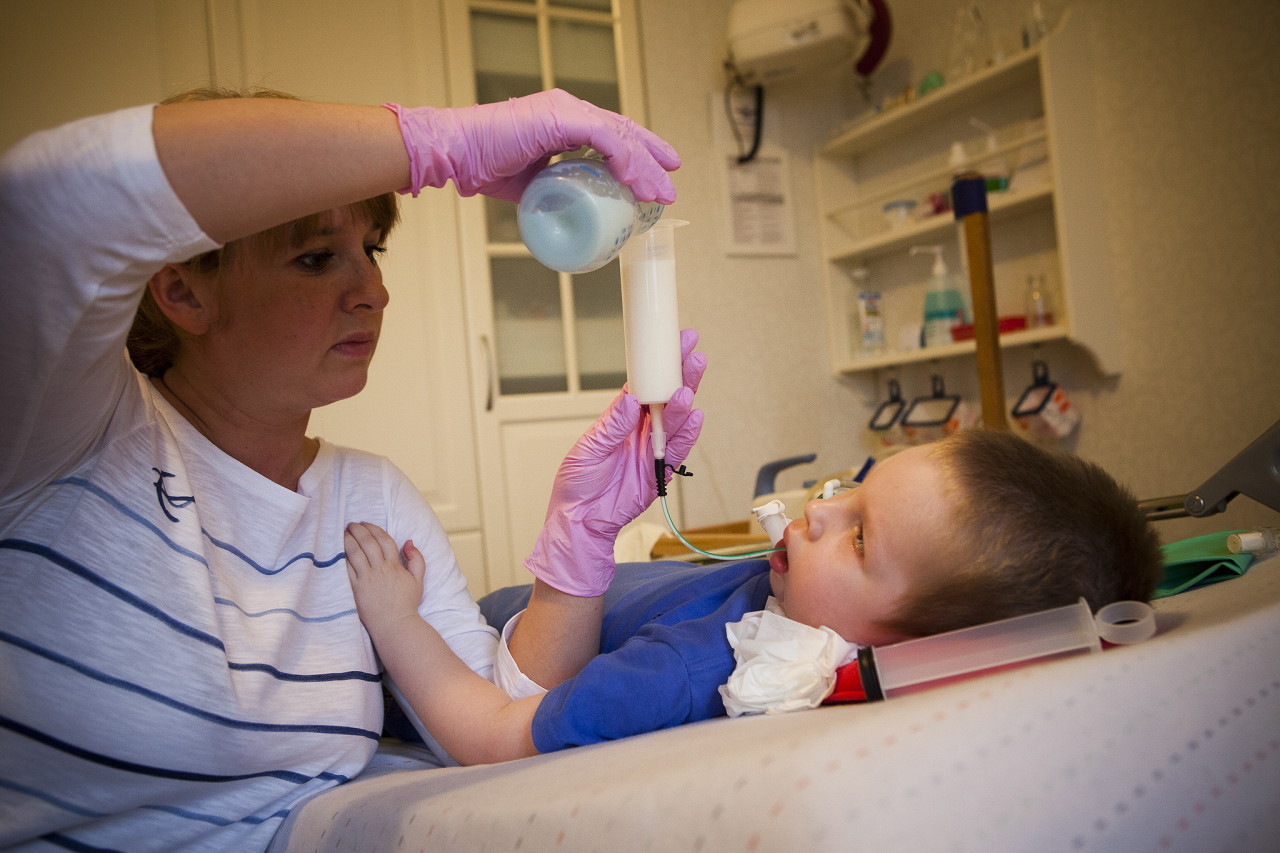
Ayudar a extender el período de vida más o menos satisfactoria ayudará. Masaje terapéutico, UHF, electroforesis, ejercicios de respiración para mantener los músculos respiratorios, natación. Se recomienda el uso de aparatos ortopédicos espinales y torácicos de soporte.

Más información sobre la enfermedad le dice a un especialista en el video a continuación.